
Abstract
The Val122Ile mutation in Transthyretin (TTR) gene causes a rare, difficult to diagnose hereditary form of cardiac amyloidosis. This mutation is most common in the United States and mainly present in people of African descent. The carriers have an increased risk of congestive heart failure and several other non-cardiac phenotypes such as carpal tunnel syndrome, peripheral edema, and arthroplasty which are top reasons for ambulatory/outpatient surgeries in the country. We conducted first-ever epigenome-wide association study in Val122Ile carriers of African descent for heart disease (HD) and multiple outpatient surgeries (OS) - an early disease indicator. Five differentially methylated sites (p≤2.1e-08) in genes – FAM129B, SKI, WDR27, GLS, and an intergenic site near RP11-550A5.2 and one differentially methylated region containing KCNA6 and GALNT3 (p=1.1e-12) were associated with HD. For OS, we observe four sites – two sites in UBE2E3 and SEC14L5, and other two in intergenic regions (p≤1.8e-07) and three regions overlapping SH3D21, EVA1B, LTB4R2 and CIDEB (p≤3.9e-07). Functional PPI module analysis identified ABCA1 (p=0.001) for HS. Six cis-mQTLs were associated with one of the significant CpG sites (FAM129B; p=4.1e-24). We replicated two CpG sites (cg18546846 and cg06641417; p<0.05) in an external cohort of biopsy-confirmed cases of TTR amyloidosis. The genes identified are involved in transport and clearance of amyloid deposits (GLS, ABCA1, FAM129B); cardiac fibrosis (SKI); and muscle tissue regulation (SKI, FAM129B). These findings highlight the link between a complex amyloid circuit and diverse symptoms of Val122Ile.
1. Introduction
Hereditary transthyretin amyloidosis, caused by specific disease-causing mutations, is due to a gradual extracellular deposition of amyloid in multiple tissues primarily leading to several clinical signs and symptoms1. There are 113 known mutations in the transthyretin (TTR) gene2 giving rise to hereditary form of TTR amyloidosis. The tetrameric structure of TTR protein dissociates into dimers and monomers resulting in formation of fibrils. The aggregation of fibrils leads to deposition of “amyloid-fibrils” in multiple tissues causing onset of heterogenous symptoms 2,3. Val122Ile is the most prevalent TTR gene mutation in the United States and observed in populations of African descent4. The Val122Ile is a point mutation (variant - rs76992529) resulting in substitution of isoleucine with valine at 122 position. Extensive amyloid deposition seems to resemble hypertrophic cardiomyopathy such as enlargement or wall thickening leading to heart failure and atrial fibrillation5. These symptoms often are attributed to other population-prevalent cardiovascular risk factors resulting in underestimation of the clinical penetrance of the Val122Ile6. The estimated age of onset for non-cardiac precursor phenotypes for hereditary transthyretin amyloidosis is between 30 to 40 years of age7. In two retrospective studies, carpal tunnel syndrome preceded hereditary transthyretin amyloidosis diagnosis by 9-10 years8,9. According to The Transthyretin Amyloid Outcome Survey (THAOS), several cardiac, gait, gastrointestinal, neurological and renal disorders are prevalent in Val122Ile carriers4. Parallel to these findings, another study reported phenotypes associated with TTR mutations such as atrial fibrillation, myopathy related to ventricular thickness, gastrointestinal and kidney dysfunction including nausea, vomiting, and neuromuscular dysfunction10.
Recently, we reported a combined association of heart disease history and having had 10 or more outpatient (ambulatory) surgeries with Val122Ile mutation in individuals of African descent11. One of the top reasons for ambulatory surgery in the United States is arthroplasty12, which occurs in TTR-carriers years before the expected cardiac dystrophy at advanced ages13. These epidemiological findings indicate that atypical phenotypes occurring earlier in life could be connected to the risk of heart failure in Val122Ile carriers. Findings from several studies including ours raise the possibility of non-regulatory molecular factors contributing to the genotype-phenotype correlation10,14–17. Therefore, understanding the underlying biological changes in Val122Ile carriers is key to explaining the symptom heterogeneity and earlier onset of atypical phenotypes.
DNA methylation is a heritable non-coding regulatory mechanism causing phenotypic variation16. Epigenetic modifications arising from the addition of methyl groups on cytosine-phosphate-guanosine (CpG) sites18 could contribute to molecular mechanisms involved in TTR amyloidosis. So far, no study has investigated this hypothesis. Aberrant methylation profiles have been implicated in increasing accelerating the progression of common and rare diseases19. The accumulation of amyloid-fibrils within or around cellular structures of the tissue result in damage invoking an immune response20. The inter-individual variation in response to site of damage invokes an acute phase response20. DNA methylation profiles have the potential to capture individual-level variability and highlight mechanisms involved in TTR amyloidosis21. Thus, we conducted the first epigenome-wide association study of TTR Val122Ile carriers to investigate the association of methylation changes with medical history of heart disease and outpatient surgeries.
2. Methods
2.1 Cohort description
Carriers of Val122Ile (rs76992529*G>A) risk allele were selected from the Yale-Penn cohort,22–25 whose medical history was obtained as part of the phenotyping effort, as previously reported11. The Yale-Penn study was approved by the institutional review boards at each participating site. The current study was approved under the protocol 2000023750 by the institutional review board (IRB) at Yale University School of Medicine.
We investigated two binary phenotypes – self-reported history of heart disease (“Has a doctor ever told you that you have (had) a heart disease?”) and having had 10 or more outpatient surgeries. We previously reported that these two phenotypes were associated with the Val122Ile mutation in the Yale-Penn cohort11. We assayed 104 African Americans, samples from 8 of which failed the assay, leaving 96 carriers for analysis (mean age = 42.16 ± 9.9 [SD] yrs.). Epigenomic differences were tested with respect to the history of heart disease (N=90 controls; 6 cases; Males-47%) and 10 or more outpatient surgeries (N=94 controls, 2 cases; Males-46%).
The significant methylation sites were investigated in an independent cohort of Italian participants of European descent. This study included 48 carriers of TTR mutations (Val30Met, Phe64Leu, Ile68Leu, Ala120Ser, and Val122Ile) with the diagnosis of TTR amyloidosis confirmed via positive amyloid fibril deposition in their cardiac tissue biopsy samples, in addition to clinical symptoms. Thirty-two healthy individuals with none of the TTR mutations or clinical symptoms from the same local area were recruited as controls. Detailed information regarding this cohort was previously reported14,26. The methylation array analysis was performed at the Connecting Bio-research and Industry Center, Trieste, Italy, using the same sample protocol implemented in the Yale-Penn cohort.
2.2 Sample and array processing
DNA was extracted from whole blood of Yale-Penn participants using the EZ-96 DNA methylation kit (Zymo Research, CA, USA). As previously described25, the samples were genotyped at the Yale Center for Genome Analysis (YCGA), the Center for Inherited Disease Research, and the Gelernter laboratory at Yale (VA CT) using genome-wide arrays (Illumina HumanOmni1-Quad v1.0 and Illumina HumanCoreExome arrays), the imputation was performed with IMPUTE2.0, and principal components were derived on the QC’d genomic data. The Val122Ile – rs76992529 probe was genotyped in the HumanCoreExome array and imputed in the HumanOmni1-Quad v1.0 array with high imputation quality (INFO score = 0.98) to determine TTR carriers in the African-American Yale-Penn participants11. DNA methylation was assayed using the Illumina Infinium MethylationEPIC chip quantifying >850,000 CpG sites and imaged on the Illumina iScan system at YCGA. The methylation intensity data (*.idat files) was exported for analysis using the manufacturer’s recommended protocol using the GenomeStudio methylation module.
2.3 DNA methylation analysis
All analyses were performed in R 3.6. The methylation intensity files (*.idat) were imported into ChAMP 27 for post-processing and normalization. The beta values, ranging from 0 to 1 were generated for all CpG sites representing the ratio of methylated to unmethylated fluorescent intensities. Primary QC removed CpG sites with low detection p-value, missing beads, sites near SNPs, multi-hit and non-autosomal sites. The beta values of the remaining 737,385 sites were normalized using the beta mixture quantile (BMIQ) method, followed by analysis of batch effects using singular vector decomposition. Technical batch effects for array and slide were corrected using ComBat. The blood cell-type composition and smoking status28 was derived from methylation data. The association analysis for CpG sites was performed on M-values (transformed beta values) using empirical Bayes methods implemented in the limma package. The association was adjusted for age, sex, tobacco use, smoking, genotype-derived principal components 1-10, and blood cell type proportions: CD8+T cells, CD4+ T cells, natural killer cells, B-cells, monocytes, and neutrophils. Genomic inflation was calculated using the QQperm package; both phenotypic associations had a lambda of 1 or less (Heart Disease – 1.01 and Outpatient Surgery – 0.92) indicating lack of population substructure bias. Due to the unbalanced case-control proportions, we also performed permutation of the association using the CpGassoc package29. Differential methylation of regions was calculated using DMRcate. The sites and regions were deemed significant considering a false discovery rate of <0.05 (FDRp-value<0.05). Differentially expressed PPI network modules were investigated using the FEM package30. Gene ontology using significant genes mapped from significant CpG sites and regions was assessed with ShinyGO 31. The cis-methylation QTL (association between SNPs and significant methylated CpG sites) was performed with all the aforementioned covariates using MatrixQTL32, wherein the local distance was defined using the biologically expected and default value of 1 Mb between SNP and CpG sites. Significant associations were identified using genomic control adjustment (GC) p-values < 0.05, in addition to FDRp-value < 0.05. Epigenetic age – defined as the cumulative score of specific CpG sites across the genome -- was calculated using Hannum and Horvath’s clock from the wateRmelon package33. As suggested by the developers of each of the epigenetic clocks, biological age, and chronological age were significantly correlated, R≥0.90; p<0.001. The difference in the epigenetic (biological) age between cases and controls was tested using Student’s t-test. The network connecting all of the significant genes detected from the above analyses was constructed using GeneMANIA34. The results were visualized using ggplot2, coMET35, and FUMA36.
3. Results
3.1 Differentially methylated sites
We investigated differentially methylated sites with respect to two binary outcomes: – a) self-reported heart disease and b) a history of 10 or more outpatient surgeries. After performing the recommended quality control procedure, we investigated 737,385 sites in 96 individuals. In addition to performing standard association analysis, we permuted the phenotypes (pperm: p-value from permutation), which accounted for the case-control imbalance, yielding nine significant CpG sites (Figure 1). Five sites were hypomethylated in individuals with heart disease: cg06641417 (FAM129B; logFC=-1.822; pperm=1.6e-08), cg26033908 (SKI; logFC=-1.615; pperm=1.7e-08), cg14890866 (WDR27; logFC=-2.028; pperm=3.0e-08), cg15522719 (GLS; logFC=-1.731; pperm=4.7e-08) and cg18546846 (intergenic; logFC=-0.786; pperm=2.2e-08). The CpG sites mapped to FAM129B and SKI are located in gene bodies, cg14890866 is between the 5’UTR (Un-Translated Region) and TSS200 (− 200 nt upstream of Transcription start site37) of WDR27, while cg15522719 is in TSS150 at GLS. Four methylation sites were associated with 10 or more outpatient surgeries: cg13998023 (UBE2E3; logFC=-2.632; pperm=1.8e-07), cg05189127 (intergenic; logFC=1.885; pperm=1.4e-07), cg03718655 (SEC14L5; logFC=-2.673; pperm=1.5e-07) and cg25814327 (intergenic; logFC=-2.075; pperm=3e-08). Three sites were hypomethylated, while cg05189127 (intergenic) was hypermethylated. The two sites that were annotated to genes were in the 5’UTR (UBE2E3) and TSS200 (SEC14L5). Details of the association result and annotation are reported in Supplementary file1 (Table S1).

(A) Methylation sites that were significantly associated with medical history of heart disease. (B) Methylation sites that were significantly associated for having had 10 or more outpatient surgeries. Each CpG site is represented as a data point, with the x-axis being the genomic location and the y-axis is the -log10 of the p-value of the CpG site. Significant sites are shown as triangles and labelled with genic annotation in parentheses, triangles pointing upwards signify hypermethylation, whereas triangles pointing downwards signify hypomethylation.
3.2 Differentially methylated regions
For heart disease, one region on chromosome 12 overlapping KCNA6 and GALNT3 (p=1.1e-12) was differentially methylated. Associations with more than 10 outpatient surgeries were identified on chromosome 1 (SH3D21; EVA1B; p=1.3e-09), chromosome 10 (intergenic region; p=1.7e-08) and chromosome 14 (LTB4R2; CIDEB; p=3.9e-07). Methylation levels among all sites were positively correlated within each region. (Figure 2; Supplementary file 1; Table S2)

(A) Regional association with heart disease (B-D) Regional association with 10 or more outpatient surgeries. Each panel displays the association of sites within each region, followed by genomic location, ENSEMBL gene name, DNAse, regulation, SNP tracks (from UCSC browser) and correlation of CpG sites shown as a heatmap.
3.3 Overrepresented gene ontology and PPI networks
Differentially methylated sites and regions were annotated to their respective genes using UCSC RefGene: hg19 genome build. The gene ontology (GO) analysis identified 15 significantly enriched pathways in GO’s biological process. GLS, SKI, GALNT8, and KCNA6 are involved in protein oligomerization (FDRp-value=4.8e-03) and KCNA6 and GALNT8, which are located near one another, are involved in potassium ion transport (FDRp-value=4.9e-02). FAM12B and SKI (FDRp-value=4.8e-03 to 3.2e-02) participate in the development of various tissue types – myotubules, and skeletal and striated muscles (Figure 3).

The dendrogram shows the FDRpvalue of the pathway associations and are grouped by similarity of function. The genes involved in each of the processes are highlighted in orange bars.
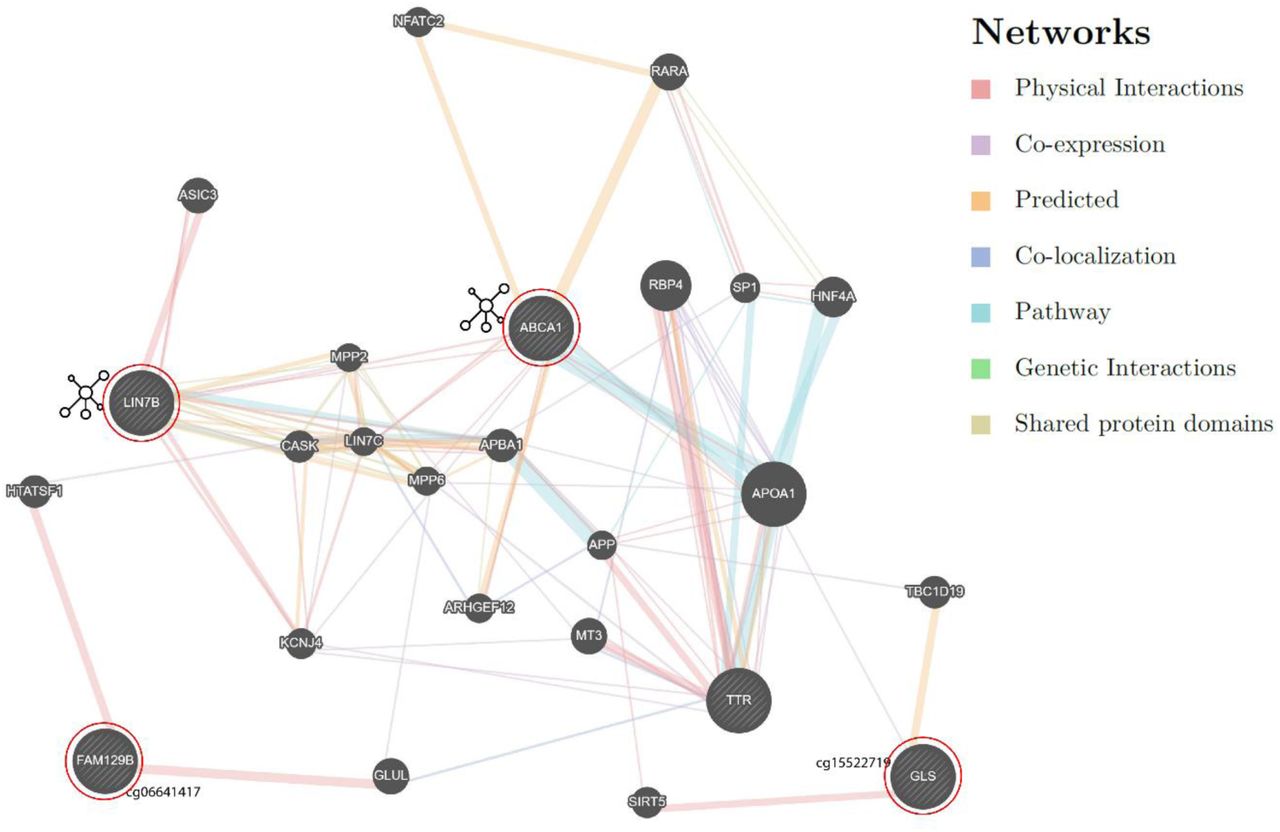
We also investigated the methylation sites for differentially methylated functional modules using the R package FEM 30. The CpG sites are weighted based on their location in the genes, which are then mapped to a protein-protein interaction (PPI) network. For each module (i.e. PPI network) identified, the seed gene is the primary gene to which other functionally related genes are connected. For heart disease, we found the ABCA1 module to be significant (p=0.001) and target genes identified within the module were: ABCA1, SNTB2, BLOC1S2 and LIN7B (p<0.05). The EXOSC4 gene module was associated with the phenotype of 10 or more outpatient surgeries, and it was the only gene that was a target (Figure 4; Supplementary file1; Table S4).

The differentially methylated modules consist of a network of genes based on their functional connectivity using protein-protein interaction. Each module has primary gene which is connected to other target genes in the network. Each module was significant p<0.05 using the FEM method (see methods). The genes in blue show hypermethylation and yellow represents hypomethylation. A) ABCA1 module was associated with heart disease and the significant target genes in addition to ABCA1 were SNTB2, BLOC1S2 and LIN7B. B) EXOSC4 module was associated with outpatient surgeries and also was the only significant gene in the network.

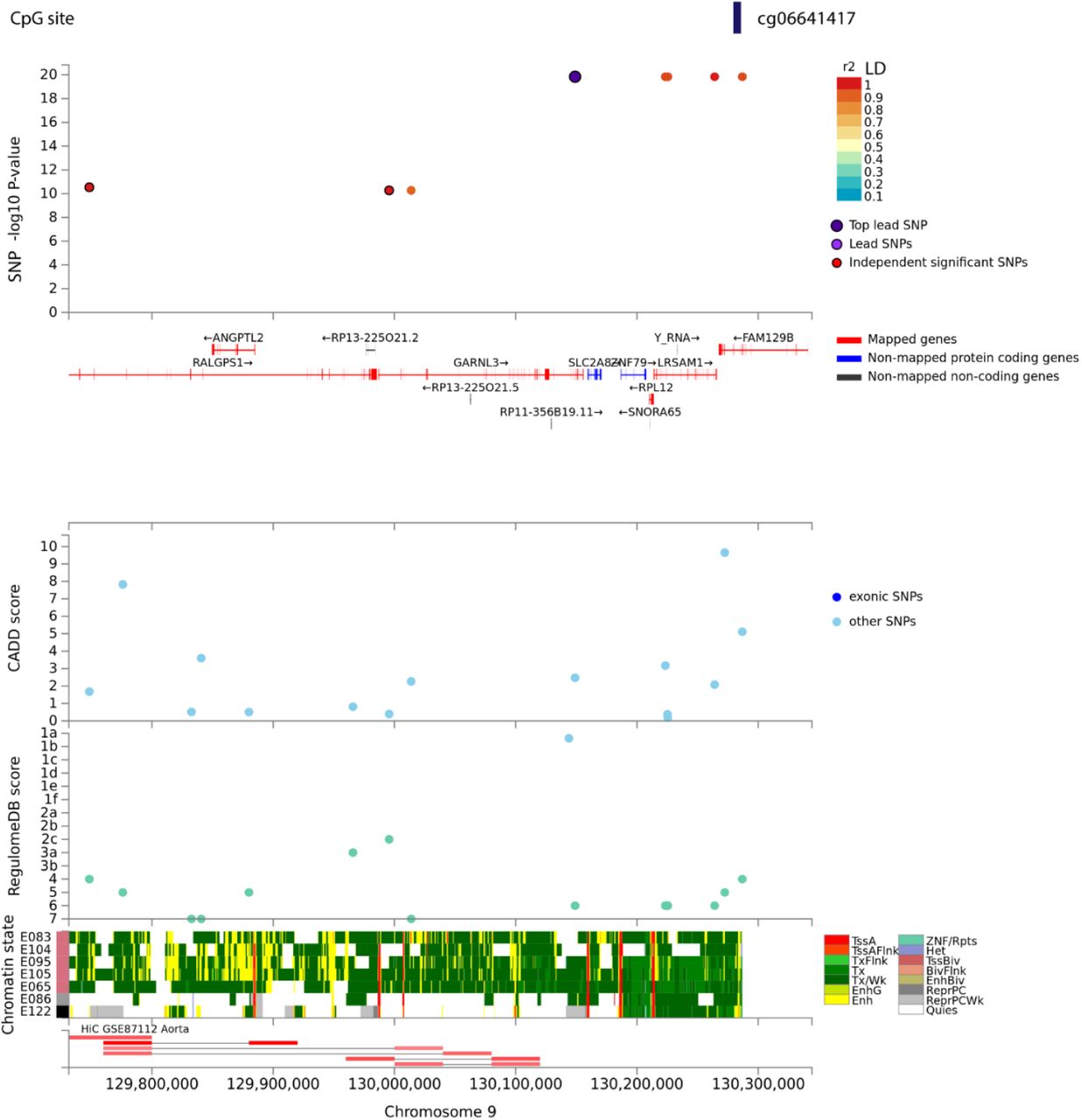
The top panel displays single nucleotide polymorphisms (SNPs) associated with CpG site – cg06641417 as a data point and color coded based on linkage disequilibrium with the top lead SNP in purple. The x-axis shows gene annotation (hg19) of the region and the y-axis displays the -log10 of p-value. The following panels present various annotations of the reported SNPs i.e. CADD - Combined Annotation Dependent Depletion, and RegulomeDB – score to identify regulatory variants. The bottom panel highlights the chromatin states of various regulatory features being putatively affected from chromatin markers observed in aorta tissue cell line. Visualization made in FUMA.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The genes (query genes) circled in red were identified from our analysis. The network shows intermediate genes that connect our query genes. Detailed information on the is provided in Supplementary file2
3.4 Local quantitative trait loci for methylated sites (mQTL)
We tested SNP associations with nine methylation sites that were epigenome-wide significant with the two phenotypes investigated. The cis-mQTL loci were defined as SNPs within ±1 Mb of the significant CpG site. The sites were considered significant based on an FDRp-value < 0.05 and genomic corrected p-value (pgc<0.05). We found six SNPs, rs192528579, rs182192023, rs114553373, rs187644239, rs114896522, and rs139996037 significantly associated (p=4.1e-24) with site cg06641417. The SNPs are in high linkage disequilibrium (LD>0.8), rs192528579 is in the intronic region of neighboring gene – GARNL3; rs182192023, rs114553373, rs187644239 and rs114896522 map to LRSAM1. Rs139996037 is a non-coding transcript variant of the FAM129B.
3.5 Epigenetic age
The epigenetic age (DNAm) was measured using the biological clock developed by Horvath and colleagues, which uses 353 CpG sites38 and also with a second clock based on 71 CpG sites from Hannum and colleagues39. The ‘Horvath’ clock is considered to be a pan-tissue epigenetic clock, while the ‘Hannum’ clock is considered to be accurate for whole-blood tissue40. Both clocks estimated that carriers with heart disease are of older epigenetic age than carriers without heart disease (pHorvath=0.007 and pHannum=0.0009) (Supplementary file1; Table S6).
3.6 Replication of methylation sites in the Italian cohort
We tested the nine CpG sites identified in Val122Ile carriers in an independent cohort of biopsy-confirmed TTR amyloidosis cases and healthy controls. We replicated cg18546846 (intergenic; near to RP11-550A5.2; p= 0.021) and cg06641417 (FAM129B; p=0.016) at nominal significance (p<0.05).
4. Discussion
The clinical consequences of the TTR Val122Ile mutation remain underappreciated and the syndrome that accompanies this risk mutation, under-diagnosed. Individuals exhibiting early TTR-amyloidosis symptoms are more likely to be diagnosed with another condition prior to receiving the diagnosis of TTR-amyloidosis41. There is nonetheless a greater burden over time towards developing ventricular hypertrophy, reduced left ventricular ejection fraction, and atrial dilation, at a later age3,5. We previously showed that African-American carriers of the Val122Ile mutation had a higher prevalence of heart disease and having multiple outpatient surgeries than individuals without the mutation11. In the present study, we identified methylation changes associated with these same phenotypes in Val122Ile carriers. We also replicated two of our CpG sites near RP11-550A5.2 and in FAM129B at nominal significance in a cohort that included confirmed cases of TTR amyloidosis. Thus, we hypothesize that the epigenetic changes associated with the pathogenesis heart disease differs from the methylation profile of carriers who are not affected by the disease. Lastly, we used GeneMANIA34 to interpret the interaction among the significant genes (Supplementary file2). We observed that major genes identified in the present study physically interact and share pathways with TTR.
ABCA1 (ATP binding cassette transporter A1) identified via the functional network analysis encodes a transporter of cholesterol from apolipoproteins42. ABCA1 regulates Apolipoprotein E (ApoE) levels, with lower expression of ABCA1 reducing ApoE levels. However, ApoE with ApoA1 (Apolipoprotein A) reduces amyloid deposition twice as fast as inhibition of the expression of ApoE. Additionally, amyloid-beta levels were the lowest for the dual-knockout of APP (which encodes amyloid precursor protein) and ABCA143.
GLS (glutaminase) is a key contributor to the metabolizing of glutamine to glutamate44. Amyloid-beta-treated neurons show elevated glutaminase expression, which increases glutamate levels and disrupts calcium neural regulation45. Additionally, neurofibrillary tangles have been shown to coexist with higher glutaminase activity46. The hypomethylated site in the transcription start site of the GLS gene may indicate its potential involvement in the central nervous system, which supports the recent finding of cerebral amyloid angiopathy in individuals with mutated TTR cardiac amyloidosis47. FAM129B (aliases; MEG-3 and NIBAN2) is downregulated in tissues with amyloid deposition and animal studies have shown that enhancing the expression of FAM129B reduces oxidative damage by reducing amyloid-beta production via PI3K/Akt signaling48. Cardiac hypertrophy increases the risk of heart failure. FAM129B is overexpressed in heart failure samples, and rodent experiments indicate a potential role of the gene in the apoptosis of cardiac myocytes after myocardial infarction49. Rescuing the expression levels of FAM129B reverses hypertrophic responses, thus the hypomethylation of the CpG site in FAM129B observed in our finding supports the overexpression of the gene in heart failure50. The African-American population has a high prevalence of diabetes51. FAM129B is also overexpressed in cardiomyocytes under high glucose concentration reflecting its role in diabetic cardiomyopathy52. Although SKI is an inhibitor of TGF-beta-induced fibrosis and is under expressed in cardiac fibrosis, other epigenetic modulators such as miRNAs-34a and 93-c affect both SKI and TGF-beta, targeting the inhibitory factors of SKI, which could rescue cardiac fibrosis53. The gene enrichment analysis identified a role for FAM129B and SKI in the development of myotube cells and skeletal muscle fiber and organ, and striated muscle cell development.
One of the clinical findings associated with cardiac amyloidosis is increased left ventricular wall thickness, which can lead to heart failure54. Electrical perturbations resulting from lower potassium repolarizing current leads to a prolonged action potential in heart failure55. The role of KCNA6 and GALNT8 is associated with potassium ion transport and the transmembrane transport complexes. One of the cardiovascular symptoms of the TTR amyloidosis is pronounced diastolic hypertension6, and diastolic dysfunction is one of the symptoms associated with transthyretin amyloidosis56. WDR27 was reported to be differentially methylated in individuals with significant differences in diastolic blood pressure26.
Aging is a common denominator to the symptomology of Val122Ile and DNA methylation57. Age-related methylation changes measured via “epigenetic clocks” help to identify molecular aging and its disconnect with chronological age. The Horvath clock based on 353 CpG sites and the Hannum clock based on 71 CpG sites have been extensively replicated in various tissues58. While these clocks were developed using blood tissues38,39, Horvath’s clock is validated across multiple tissues, while Hannum’s clock is more consistent in samples originating from blood tissues. Higher epigenetically derived age has been associated with several cardiovascular disease traits. Hypermethylation of genes that are protective against heart disease, lead to cardiovascular aging and increased risk for coronary disease59. The dysregulation of the ABCA1 gene, the product of which is involved in the transport of cholesterol from the periphery to liver tissue60 has been associated with different cardiovascular pathologies. The hypermethylation of the ABCA1 promoter region silences its expression and is associated with coronary artery disease61. In contrast, the increased expression of ABCA1 regulated by ApoA1 leads to reverse cholesterol efflux in hepatic tissue 62. Elevated high density lipoprotein (HDL) in the liver is a target site for serum amyloid A, an acute phase response protein that is expressed during amyloidosis63. The observed hypomethylation of ABCA1 and putative increase in gene expression underscores its likely involvement in shifting the methylation milieu and could perhaps explain the cardiac symptomology in a comparatively younger group of Val122Ile carriers.
These findings provide unique insights into epigenomic contrasts related to symptomology in Val122Ile carriers. However, our study has limitations. First, we investigated the Val122Ile polymorphism only for heart disease, though it is possible that we could identify additional differences with individuals who are non-Val122Ile carriers or who present with wild-type transthyretin amyloidosis. Additionally, due to the low frequency of the disease-causing mutation investigated, our study suffers from an imbalance in the ratio of cases to controls. Although the permutation analysis accounting for this imbalance confirmed our results and we replicated two associations in an independent cohort (with mostly different risk variants), our findings would benefit from replication in a larger, more balanced study.
5. Conclusion
Our study is the first to explore the epigenetic changes in TTR Val122Ile carriers. Certain Val122Ile carriers in our study presented with heart disease earlier than usually reported by individuals affected by cardiac amyloidosis. This could be due to modifier effects accelerating the pathogenicity of Val122Ile mutation. Due to the underestimated clinical penetrance of the mutation in the African American population, we leveraged an external secondary dataset with confirmed clinical phenotype as a random population sample. The purpose of this study was to understand possible non-coding mechanisms that may explain the heterogeneous phenotypes observed in Val122Ile carriers with history of heart disease. The epigenetic changes identified affect the regulation of genes involved in the transport of amyloid and regulating striated and smooth muscle, which form key components of amyloidosis and cardiac tissue susceptibility. These findings provide higher resolution on mechanisms underlying the TTR-Val122Ile mutation.
Data Availability
Full summary statistics of the epigenome-wide analysis will be made available post-publication by contacting the corresponding author – R.P. listed on the manuscript.
7. Author contributions
R.P. and G.A.P. conceptualized the research design and R.P. received funding for the study. J.G. and H. K designed the Yale-Penn study, and H.K. and J.G. oversaw the recruitment and assessment of the Yale-Penn sample. The analysis was performed by G.A.P. All authors were involved in data interpretation, manuscript preparation, review, and critical feedback. The funding for Yale-Penn was obtained by J.G and H.K.
8. Conflicts of Interest
H.R.K. is a member of the American Society of Clinical Psychopharmacology’s Alcohol Clinical Trials Initiative, which over the last three years was sponsored by Alkermes, Ethypharm, Indivior, Lilly, Lundbeck, Otsuka, Pfizer, Arbor Pharmaceuticals, and Amygdala Neurosciences, Inc. H.R.K. and J.G. are named as inventors on PCT patent application #15/878,640 entitled: “Genotype-guided dosing of opioid agonists,” filed on 24 January 2018. The other authors report no conflict of interest.
6. Acknowledgments
The study was supported by ‘Global ASPIRE TTR Amyloidosis Competitive Grant’ from Pfizer Inc. We are grateful to the participants of the Yale-Penn cohort, which was funded under grants RC2 DA028909, R01 DA12690, R01 DA12849, R01 DA18432, R01AA11330, and R01 AA017535. The investigation conducted in the Italian cohort was supported by an Investigator-Initiated Research from Pfizer Inc. to the University of Rome “Tor Vergata”. The content reported in the manuscript is solely the responsibility of the authors and does not represent the official views of the NIH or Pfizer. The funding agencies had no role in the study design, data analysis, and results interpretation of the present study.
Full summary statistics of the epigenome-wide analysis will be made available post-publication by contacting the corresponding author – R.P. listed on the manuscript.
9. References




