
ABSTRACT
We probe how genetic variability across the three major histocompatibility complex (MHC) class I genes (human leukocyte antigen [HLA] A, B, and C) may affect susceptibility to and severity of severe acute respiratory syndrome 2 (SARS-CoV-2), the virus responsible for coronavirus disease 2019 (COVID-19). We execute a comprehensive in silico analysis of viral peptide-MHC class I binding affinity across all known HLA -A, -B, and -C genotypes for all SARS-CoV-2 peptides. We further explore the potential for cross-protective immunity conferred by prior exposure to four common human coronaviruses. The SARS-CoV-2 proteome is successfully sampled and presented by a diversity of HLA alleles. However, we found that HLA-B*46:01 had the fewest predicted binding peptides for SARS-CoV-2, suggesting individuals with this allele may be particularly vulnerable to COVID-19, as they were previously shown to be for SARS (1). Conversely, we found that HLA-B*15:03 showed the greatest capacity to present highly conserved SARS-CoV-2 peptides that are shared among common human coronaviruses, suggesting it could enable cross-protective T-cell based immunity. Finally, we report global distributions of HLA types with potential epidemiological ramifications in the setting of the current pandemic.
IMPORTANCE Part of why the immune response to a virus differs across individuals is explained by genetic variation, especially in the HLA genes. Understanding how variation in HLA may affect the course of COVID-19 could help identify individuals at higher risk from the disease. HLA typing can be fast and inexpensive. Pairing HLA typing with COVID-19 testing where feasible could thus improve assessment of viral severity in the population. In the event of the development of a vaccine against SARS-CoV-2, the virus that causes COVID-19, high-risk individuals could be prioritized for vaccination.
INTRODUCTION
Recently, a new strain of betacoronavirus (severe acute respiratory syndrome coronavirus 2, or SARS-CoV-2) has emerged as a global pathogen, prompting the World Health Organization in January 2020 to declare an international public health emergency (2). In the large coronavirus family, comprising enveloped positive-strand RNA viruses, SARS-CoV-2 is the seventh encountered strain that causes respiratory disease in humans (3) ranging from mild -- the common cold -- to severe -- the zoonotic Middle East Respiratory Syndrome (MERS-CoV) and Severe Acute Respiratory Syndrome (SARS-CoV). As of March 2020, there are in excess of 340,000 presumed or confirmed cases of coronavirus disease 19 (COVID-19) worldwide, with total deaths exceeding 14,000 (4). While age and many comorbid health conditions, including cardiovascular and pulmonary disease, appear to increase the severity and mortality of COVID-19 (5–10), approximately 80% of infected individuals have mild symptoms (11). As with SARS-CoV (12, 13) and MERS-CoV (14, 15), children in particular seem to have low susceptibility to the disease (16–18); despite similar infection rates as adults (19) only 5.9% of pediatric cases are severe or critical, possibly due to lower binding ability of the ACE2 receptor in children or generally higher levels of antiviral antibodies (20). Other similarities (21–23) including genomic (24, 25) and immune system response (26–34) between SARS-CoV-2 and other coronaviruses (35), especially SARS-CoV and MERS-CoV, are topics of ongoing active research, results of which may inform an understanding of the severity of infection (36) and improve the ongoing work of immune landscape profiling (37–41) and vaccine discovery (30, 39, 42–48).
Previous studies of other viruses have shown that genetic variability among human leukocyte antigen (HLA) alleles, which are critical components of the viral antigen presentation pathway, may confer differential viral susceptibility and influence disease severity. For instance, disease caused by the closely-related SARS-CoV (24, 25) shows increased severity among individuals with the HLA-B*46:01 genotype (1). Associations between HLA genotype and disease severity extend broadly to several other unrelated viruses including human immunodeficiency virus 1 (HIV-1), where certain HLA types (e.g. HLA-A*02:05) may reduce risk of seroconversion (49), and dengue virus, where certain HLA alleles (e.g. HLA-A*02:07, HLA-B*51) are associated with increased secondary disease severity among ethnic Thais (50).
While a detailed clinical picture of the COVID-19 pandemic continues to emerge, there remain substantial unanswered questions regarding the role of individual genetic variability in the immune response against SARS-CoV-2. We hypothesize that individual HLA genotypes may differentially induce the T-cell mediated antiviral response, and could potentially alter the course of disease and its transmission. In this study, we performed a comprehensive in silico analysis of viral peptide-MHC class I binding affinity, across 145 different HLA types, for the entire SARS-CoV-2 proteome.
RESULTS
To explore the potential for a given HLA allele to produce an antiviral response, we assessed the HLA binding affinity of all possible 8-12mers from the SARS-CoV-2 proteome (n=48,395 unique peptides). We then removed from further consideration 16,138 peptides that were not predicted to enter the MHC class I antigen processing pathway via proteasomal cleavage. For the remaining 32,257 peptides, we repeated binding affinity predictions for a total of 145 different HLA types, and we show here the SARS-CoV-2-specific distribution of per-allele proteome presentation (predicted binding affinity threshold <500nM, Figure 1, Supplementary Table S1). Importantly, we note that the putative capacity for SARS-CoV-2 antigen presentation is unrelated to the HLA allelic incidence in the population (Figure 1). We identify HLA-B*46:01 as the HLA allele with the fewest predicted binding peptides for SARS-CoV-2. We performed the same analyses for the closely-related SARS-CoV proteome (Supplementary Figure S1) and similarly note that HLA-B*46:01 was predicted to present the fewest SARS-CoV peptides, in keeping with previous clinical data associating this allele with severe disease (1).

Distribution of HLA allelic presentation of 8-12mers from the SARS-CoV-2 proteome. At right, the number of peptides (see Supplementary Table S1) that putatively bind to each of 145 HLA alleles is shown as a series of horizontal bars, with dark and light shading indicating the number of tightly (<50nM) and loosely (<500nM) binding peptides respectively, and with green, orange, and purple colors representing HLA-A, -B, and -C alleles, respectively. Alleles are sorted in descending order based on the number of peptides they bind (<500nM). The corresponding estimated allelic frequency in the global population is also shown (to left), with length of horizontal bar indicating absolute frequency in the population.
To assess the potential for cross-protective immunity conferred by prior exposure to common human coronaviruses (i.e. HKU1, OC43, NL63, and 229E), we next sought to characterize the conservation of the SARS-CoV-2 proteome across diverse coronavirus subgenera to identify highly conserved linear epitopes. After aligning reference proteome sequence data for 5 essential viral components (ORF1ab, S, E, M, and N proteins, which together comprise the vast majority of the SARS-CoV-2 proteome) across 34 distinct alpha- and betacoronaviruses, including all known human coronaviruses, we identified 48 highly conserved amino acid sequence spans (Appendix 1). Among these conserved sequences, 44 SARS-CoV-2 sequences would each be anticipated to generate at least one component 8-12mer linear peptide epitope also present within at least one other common human coronavirus (Supplementary Table S2, Figure 2). In total, 564 such 8-12mer peptides were found to share 100% identity with corresponding OC43, HKU1, NL63, and 229E sequences (467, 460, 179, and 157 peptides, respectively) (Supplementary Table S3).

Amino acid sequence conservation of four linear peptide example sequences from 3 human coronavirus proteins. Protein sequence alignments are shown for nucleocapsid (N), membrane (M), and ORF1ab polyprotein (Helicase) across all 5 known human betacoronaviruses (SARS-CoV-2, SARS-CoV, HKU1, OC43, and MERS-CoV) and two known human alphacoronaviruses (229E and NL63). Each row in the 3 depicted sequence alignments corresponds to the protein sequence from the indicated coronavirus, with starting coordinate of the viral protein sequence shown at left and position coordinates of the overall alignment displayed above. Blue shading for each amino acid indicates the extent of sequence identity, with the darkest blue shading indicating 100% match for that amino acid across sequences. The 4 red highlighted sequences correspond to highly conserved peptides ≥8 amino acids in length (PRWYFYYLGTGP, WSFNPETN, QPPGTGKSH, VYTACSHAAVDALCEKA, see Supplementary Table S2).
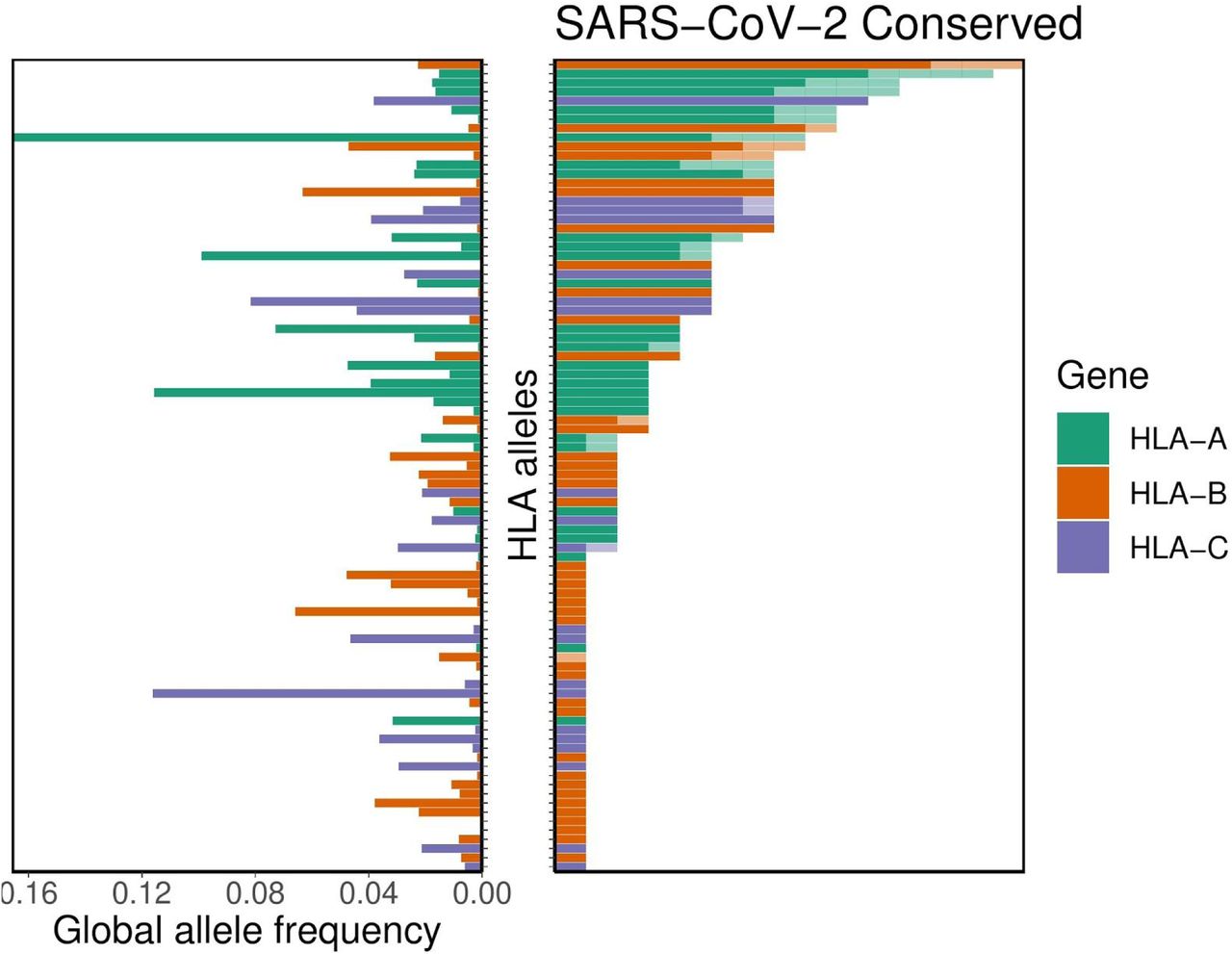
For the subset of these potentially cross-protective peptides that are anticipated to be generated via the MHC class I antigen processing pathway, we performed binding affinity predictions across 145 different HLA-A, -B, and -C alleles. As above, we demonstrate the SARS-CoV-2-specific distribution of per-allele presentation for these conserved peptides. We found that alleles HLA-A*02:02, HLA-B*15:03, and HLA-C*12:03 were the top presenters of conserved peptides. Conversely, we note that 56 different HLA alleles demonstrated no appreciable binding affinity (<500nM) to any of the conserved SARS-CoV-2 peptides, suggesting a concomitant lack of potential for cross-protective immunity from other human coronaviruses. We note in particular HLA-B*46:01 is among these alleles. Note also that the putative capacity for conserved peptide presentation is unrelated to the HLA allelic incidence in the population (Figure 3). Moreover, we see no appreciable global correlation between conservation of the SARS-CoV-2 proteome and its predicted MHC binding affinity, suggesting a lack of selective pressure for or against the capacity to present coronavirus epitopes (p=0.27 [Fisher’s exact test], Supplementary Figure S2).

Distribution of HLA allelic presentation of highly conserved human coronavirus peptides with potential to elicit cross-protective immunity to COVID-19. At right, the number of conserved peptides (see Supplementary Table S3) that putatively bind to a subset of 89 HLA alleles is shown as a series of horizontal bars, with dark and light shading indicating the number of tightly (<50nM) and loosely (<500nM) binding peptides respectively, and with green, orange, and purple colors representing HLA-A, -B, and -C alleles, respectively. Alleles are sorted in descending order based on the number of peptides they are anticipated to present (binding affinity <500nM). The corresponding allelic frequency in the global population is also shown (to left), with length of horizontal bar indicating absolute frequency in the population.
We were further interested in whether certain regions of the SARS-CoV-2 proteome showed differential presentation by the MHC class I pathway. Accordingly, we surveyed the distribution of antigen presentation capacity across the entire proteome, highlighting its most conserved peptide sequences (Figure 4). Throughout the entire proteome, HLA-A and HLA-C alleles exhibited the relatively most and least capacity to present SARS-CoV-2 antigens, respectively. However each of the three major class I genes exhibited a very similar pattern of peptide presentation across the proteome (Supplementary Figure S3).

Distribution of allelic presentation of conserved 8-12mers across the entire SARS-CoV-2 proteome for all HLA alleles and individually for HLA-A, HLA-B, and HLA-C (first, second, third, and fourth plots from top, respectively) with dark and light shading indicating the number of tightly (<50nM) and loosely (<500nM) binding peptides, respectively. Positions are derived from a concatenation of coding sequences (CDSes) indicated in the bottom panel. Tightly binding peptides are confined to ORF1ab. The sequence begins with only the last 12 amino acids of ORF1a because all but the last four amino acids of ORF1a are contained in ORF1ab, and we considered binding peptides up to 12 amino acids in length.
Finally, given the global nature of the current COVID-19 pandemic, we sought to describe population-level distributions of the HLA alleles best (and least) able to generate a repertoire of SARS-CoV-2 epitopes in support of a T-cell based immune response. While we present global maps of individual HLA allele frequencies for the full set of 145 different alleles studied herein (Appendix 2), we specifically highlight the global distributions of the three best-presenting (A*02:02, B*15:03, C*12:03) and three of the worst-presenting (A*25:01, B*46:01, C*01:02) HLA-A, -B, and -C alleles (Figure 5). Note that all allelic frequencies are aggregated by country, but implicitly reflect the distribution of HLA data available on the Allele Frequency Net Database (51).

Global HLA allele frequency distribution heatmaps for six HLA-A, -B, and -C alleles. The leftmost panels show the global allele frequency distributions by country for three representative alleles (HLA-A*02:02, HLA-B*15:03, and HLA-C*12:03) with the predicted capacities to present the greatest repertoire of epitopes from the SARS-CoV-2 proteome. Conversely, the rightmost panels show the global allele frequency distributions by country for three representative alleles (HLA-A*25:01, HLA-B*46:01, and HLA-C*01:02) with the least predicted epitope presentation from the SARS-CoV-2 proteome. Heatmap color corresponds to the individual HLA allele frequency within each country ranging from least (white/yellow) to most (red) frequent as indicated in the legend below each map.
DISCUSSION
To the best of our knowledge, this is the first study to evaluate per-allele viral proteome presentation across a wide range of HLA alleles using MHC-peptide binding affinity predictors. This study also introduces the relationship between coronavirus sequence conservation and MHC class I antigen presentation. We show that assessing MHC-peptide binding affinity from generated viral peptides can recapitulate previous clinical data that associates a specific allele, HLA-B*46:01, with the severity of SARS-CoV infection (1). We demonstrate that this allele also exhibits poor binding affinity with the SARS-CoV-2 proteome, suggesting that it could be similarly associated with higher COVID-19 severity. Further, we show that across 145 HLA alleles, the ability of an allele to bind is highly similar between SARS-CoV peptides and SARS-CoV-2 peptide. We found that in general, there is no correlation between the allelic frequency in the population and allelic capacity to bind SARS-CoV or SARS-CoV-2 peptides.
We note, however, several limitations to this work. As we are unable to obtain individual-level HLA typing and clinical outcomes data for any real-world COVID-19 populations, the data presented is theoretical in nature, and subject to many of the same limitations implicit to the MHC binding affinity prediction tool(s) upon which it is based. We further note that peptide-MHC binding affinity is limited as a predictor of subsequent T-cell responses (52–54). Moreover, we did not assess genotypic heterogeneity or in vivo evolution of SARS-CoV-2, which could modify the repertoire of viral epitopes presented, or otherwise modulate virulence in an HLA-independent manner (55, 56) (https://nextstrain.org/ncov). We also do not address the potential for individual-level genetic variation in other proteins (e.g. angiotensin converting enzyme 2 [ACE2] or transmembrane serine protease 2 [TMPRSS2], essential host proteins for SARS-CoV-2 priming and cell entry (57) to modulate the host-pathogen interface.
Unless and until the findings we present here are clinically validated, they should not be employed for any clinical purposes. However, we do at this juncture recommend integrating HLA testing into clinical trials and pairing HLA typing with COVID-19 testing where feasible to more rapidly develop and deploy predictor(s) of viral severity in the population, and potentially to tailor future vaccination strategies to genotypically at-risk populations. This approach may have additional implications for the management of a broad array of other viruses.
MATERIALS AND METHODS
Sequence retrieval and alignments
Full polyprotein 1ab (ORF1ab), spike (S) protein, membrane (M) protein, envelope (E) protein, and nucleocapsid (N) protein sequences were obtained for each of 34 distinct but representative alpha and betacoronaviruses from a broad genus and subgenus distribution, including all known human coronaviruses (i.e. SARS-CoV, SARS-CoV-2, MERS-CoV, HKU1, OC43, NL63, and 229E). FASTA-formatted protein sequence data (full accession number list available in Supplementary Table S4) was retrieved from the National Center of Biotechnology Information (NCBI) (58). For each protein class (i.e. ORF1ab, S, M, E, N), all 34 coronavirus sequences were aligned using the Clustal Omega v1.2.4 multisequence aligner tool employing the following parameters: sequence type [Protein], output alignment format [clustal_num], dealign [false], mBed-like clustering guide-tree [true], mBed-like clustering iteration [true], number of combined iterations [0], maximum guide tree iterations [-1], and maximum HMM iterations [-1] (59).
Conserved peptide assessment
Aligned sequences were imported into Jalview v. 2.1.1 (60) with automated generation of the following alignment annotations: 1) sequence consensus, calculated as the percentage of the modal residue per column,sequence conservation (0-11), measured as a numerical index reflecting conservation of amino acid physico-chemical properties in the alignment, 3) alignment quality (0-1), measured as a normalized sum of BLOSUM62 ratios for all residues at each position, 4) occupancy, calculated as the number of aligned residues (not including gaps) for each position. In all cases, sequence conservation was assessed for each of three groups: only human coronaviruses (n=7), all betacoronaviruses (n=16), and combined alpha- and betacoronavirus sequences (n=34). Aligned SARS-CoV-2 sequence and all annotations were manually exported for subsequent analysis. Conserved human coronavirus peptides were defined as those with a length ≥8 consecutive amino acids, each with an agreement of SARS-CoV-2 and ≥4 other human coronavirus sequences with the consensus sequence (Supplementary Table S2). For each of these conserved peptides, we also assessed the component number of 8-12mers sharing identical amino acid sequence between SARS-CoV-2 and each of the four other common human coronaviruses (i.e. OC43, HKU1, NL63, 229E) (Supplementary Table S3). For all peptides, human, beta, and combined conservation scores were obtained using a custom R v.3.6.2 script as the mean sequence conservation (minus gap penalties where relevant) (see https://github.com/pdxgx/covid19).
Peptide-MHC class I binding affinity predictions
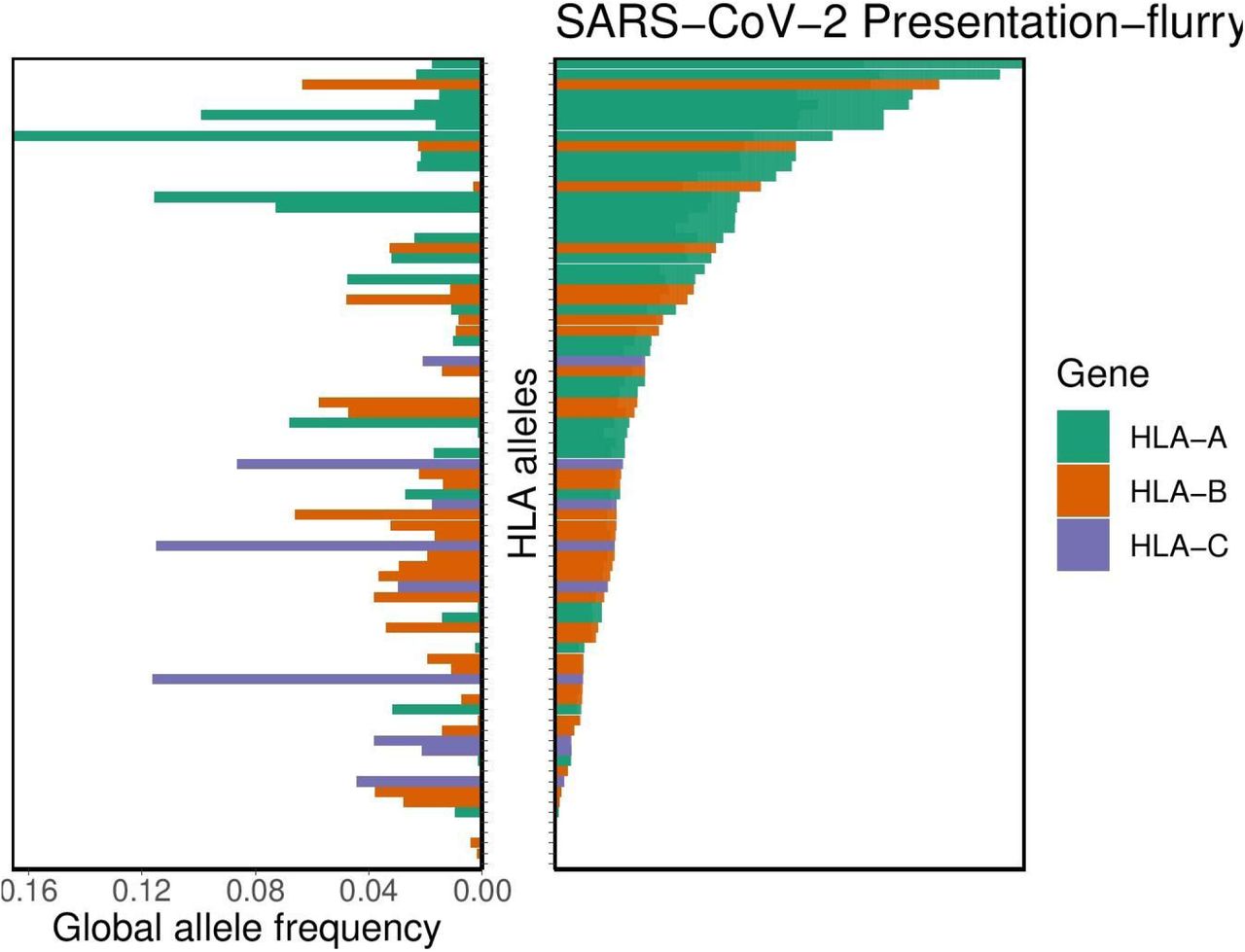
FASTA-formatted input protein sequences from the entire SARS-CoV-2 and SARS-CoV proteomes were obtained from NCBI RefSeq database (58) under accession numbers NC_045512.2 and NC_004718.3. We kmerized each of these sequences into 8-12mers to assess MHC class I-peptide binding affinity across the entire proteome. MHC class I binding affinity predictions were performed using 145 different HLA alleles for which global allele frequency data was available as described previously (61) (see Supplementary Table S1) with netMHCpan v4.0 (62) using the ‘-BA’ option to include binding affinity predictions and the ‘-l’ option to specify peptides of lengths 8-12. Binding affinity was not predicted for peptides containing the character ‘|’ in their sequences. Additional MHC class I binding affinity predictions were performed on all 66 MHCflurry supported alleles (--list-supported-alleles) using both MHCnuggets 2.3.2 (63) and MHCflurry 1.4.3 (64) (Supplementary Tables S7 and S8). We then performed consensus binding affinity predictions for the 66 supported alleles shared by all three tools by taking the union set of alleles and filtering for peptide-allele pairs matching the union set of alleles. For the SARS-CoV and SARS-CoV-2 specific distribution of per-allele proteome presentation, we exclude all peptides-allele pairs with >500nM predicted binding. In all cases, we used the netchop v3.0 (65) “C-term” model with a cleavage threshold of 0.1 to further remove any peptides that were not predicted to undergo canonical MHC class I antigen processing via proteasomal cleavage (of the peptide’s C-terminus).
Global HLA allele frequencies
HLA-A, -B, and -C allele frequency data were obtained from the Allele Frequency Net Database (51) for 805 distinct populations pertaining to 101 different countries and 2628 distinct major/minor (4-digit) alleles (see GithubXXX). Population allele frequency data was aggregated by country as a mean of all constituent population allele frequencies weighted by sample size of the population, but not accounting for representative ethnic demographic size of the population. Global allele frequency maps were generated using the rworldmap v1.3-6 package (66), with total global allele frequency estimates calculated as the mean of per-country allele frequencies, weighted by each country’s population in 2005.
Data Availability
Data and code available at https://github.com/pdxgx/covid19. Raw sequence data available through https://www.ncbi.nlm.nih.gov/refseq/.
APPENDICES
Appendix 1 contains graphical depictions of sequence alignments for 5 protein sequences (ORF1ab polyprotein, spike, envelope, membrane, and nucleocapsid) across 34 diverse coronavirus proteomes (including all 7 human coronaviruses). Appendix 2 contains a series of world maps displaying the estimated global distributions and frequencies across 145 different HLA alleles. Appendix 3 contains Supplementary Tables S1-S8 as follows: list of SARS-CoV-2 peptides and their predicted binding affinity across 145 HLA alleles (Supplementary Table S1), SARS-CoV-2 peptides conserved across diverse coronavirus sequences (Supplementary Table S2), presence of 8-12mer peptides across four human coronavirus sequences (Supplementary Table S3), coronavirus taxonomy and sequence accession numbers for conserved coronavirus proteins (Supplementary Table S4), list of 145 HLA alleles used for binding affinity predictions (Supplementary Table S5), list of SARS-CoV peptides and their predicted binding affinity across 145 HLA alleles (Supplementary Table S6), list of SARS-CoV-2 peptides and their predicted MHCnuggets and MHCflurry binding affinities across 145 HLA alleles (Supplementary Table S7), list of SARS-CoV peptides and their predicted MHCnuggets and MHCflurry binding affinities across 145 HLA alleles (Supplementary Table S8). Appendix 4 contains the full list of SARS-CoV-2 8-12mers with their individual netMHCpan v4.0 predicted binding affinities across all 145 HLA alleles.
DISCLAIMER
The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
FUNDING
RFT was supported by the U.S. Department of Veterans Affairs under award number 1IK2CX002049-01, and the Sunlin & Priscilla Chou Foundation.
SUPPLEMENTARY DATA

Distribution of HLA allelic presentation of 8-12mers from the SARS-CoV proteome (see Supplementary Table S6). At right, the number of peptides that putatively bind to each of 145 HLA alleles is shown as a series of horizontal bars, with dark and light shading indicating the number of tightly (<50nM) and loosely (<500nM) binding peptides respectively, and with green, orange, and purple colors representing HLA-A, -B, and -C alleles, respectively. Alleles are sorted in descending order based on the number of peptides they bind (<500nM). The corresponding estimated allelic frequency in the global population is also shown (to left), with length of horizontal bar indicating absolute frequency in the population.

Relationship between predicted peptide-MHC binding affinity and peptide conservation across coronaviruses. Every point represents a single unique peptide covering, together, the entirety of the SARS-CoV-2 proteome. The best predicted MHC binding affinity scores across 145 different HLA alleles are shown for each peptide along the x-axis. Sequence conservation (Clustal Omega alignment score) is shown for each peptide along the y-axis.

Pairwise relationship of peptide presentation between HLA-A, -B, and -C. In the bottom left three panels, every point represents the pairwise comparison of the number of peptide-allele interactions for all position coordinates. Taken together, the position coordinates cover the entirety of the SARS-CoV-2 proteome. The top right three panels show the quantitative correlation scores between each pair of HLA type comparisons (*** indicates statistical significance).

Distribution of HLA allelic presentation of 8-12mers from the SARS-CoV-2 proteome using the tool MHCflurry. At right, the number of peptides (see Supplementary Table S7) that putatively bind to each of 66 HLA alleles is shown as a series of horizontal bars, with dark and light shading indicating the number of tightly (<50nM) and loosely (<500nM) binding peptides respectively, and with green, orange, and purple colors representing HLA-A, -B, and -C alleles, respectively. Alleles are sorted in descending order based on the number of peptides they bind (<500nM). The corresponding estimated allelic frequency in the global population is also shown (to left), with length of horizontal bar indicating absolute frequency in the population.

Distribution of HLA allelic presentation of 8-12mers from the SARS-CoV proteome using the tool MHCflurry. At right, the number of peptides (see Supplementary Table S8) that putatively bind to each of 66 HLA alleles is shown as a series of horizontal bars, with dark and light shading indicating the number of tightly (<50nM) and loosely (<500nM) binding peptides respectively, and with green, orange, and purple colors representing HLA-A, -B, and -C alleles, respectively. Alleles are sorted in descending order based on the number of peptides they bind (<500nM). The corresponding estimated allelic frequency in the global population is also shown (to left), with length of horizontal bar indicating absolute frequency in the population.

Distribution of HLA allelic presentation of 8-12mers from the SARS-CoV-2 proteome using the tool MHCnuggets. At right, the number of peptides (see Supplementary Table S7) that putatively bind to each of 66 HLA alleles is shown as a series of horizontal bars, with dark and light shading indicating the number of tightly (<50nM) and loosely (<500nM) binding peptides respectively, and with green, orange, and purple colors representing HLA-A, -B, and -C alleles, respectively. Alleles are sorted in descending order based on the number of peptides they bind (<500nM). The corresponding estimated allelic frequency in the global population is also shown (to left), with length of horizontal bar indicating absolute frequency in the population.

Distribution of HLA allelic presentation of 8-12mers from the SARS-CoV proteome using the tool MHCnuggets. At right, the number of peptides (see Supplementary Table S8) that putatively bind to each of 66 HLA alleles is shown as a series of horizontal bars, with dark and light shading indicating the number of tightly (<50nM) and loosely (<500nM) binding peptides respectively, and with green, orange, and purple colors representing HLA-A, -B, and -C alleles, respectively. Alleles are sorted in descending order based on the number of peptides they bind (<500nM). The corresponding estimated allelic frequency in the global population is also shown (to left), with length of horizontal bar indicating absolute frequency in the population.
ACKNOWLEDGEMENTS
We thank Drs. Christopher Loo and Jeffrey Barnet for their critical reading of the manuscript. We thank Drs. Jonah Sacha and Paul Spellman for their helpful discussions.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}