
Abstract
Mitochondrial translation is essential for the biogenesis of the mitochondrial oxidative phosphorylation system (OXPHOS) that synthesizes the bulk of ATP for the cell. Mutations in either mitochondrial DNA or in nuclear genes that encode mitochondrial translation factors can result in impaired OXPHOS biogenesis and mitochondrial diseases with variable clinical presentations.
Mutations in the FARS2 gene encoding the mitochondrial phenylalanyl-tRNA synthetase are commonly linked to either early-onset epileptic mitochondrial encephalopathy or spastic paraplegia. Here, we expand the genetic spectrum of FARS2-linked disease with three patients carrying novel compound heterozygous variants in the FARS2 gene and presenting with spastic tetraparesis, axial hypotonia and myoclonic epilepsy in two cases.
Introduction
Mitochondria contain their own translation system complete with a mitochondria-specific ribosome (mitoribosome) and translation factors that function like their cytosolic counterparts but are encoded by distinct nuclear genes. Intramitochondrial translation of mitochondrial DNA (mtDNA)-encoded proteins is essential for the biogenesis of four of the five multiprotein complexes that form the ATP-producing oxidative phosphorylation system (OXPHOS). Inhibition of mitochondrial translation results in isolated or multiple OXPHOS deficiencies and mitochondrial disease.
FARS2 (OMIM# 611592) encodes the mitochondrial phenylalanyl-tRNA synthetase (mtPheRS), which charges mt-tRNAPhe with phenylalanine for translation. MtPheRS contains four major domains—N-terminal, catalytic domain, a linker region (residues 290–322) and an anticodon-binding domain (ABD)—whose complex interactions and conformational changes enable it to function as a monomer during aminoacylation (1,2). To date, 31 FARS2 mutations have been linked to two major clinical presentations: a) early-onset epileptic mitochondrial encephalopathy in about two-thirds of the cases (3-11); and b) spastic paraplegia (12-14). While the former group has a typically poorer prognosis, the latter is associated with less severe disease and prolonged survival. MRI studies of patients with early-onset disease often reveal global brain atrophy, cerebellar and basal ganglia abnormalities. In the second group, spastic paraplegia is associated with weakness of the lower extremities, spasticity and difficulties walking. Intellectual disability/developmental delay may be observed in both groups. Here, we report the identification of 4 novel disease-causing variants in the FARS2 gene in three unrelated patients (Pts) presenting with spastic tetraparesis, developmental delay and myoclonic epilepsy in two cases.
Subjects and methods
Informed consent was obtained for all patients in accordance with the Declaration of Helsinki protocols and approved by local institutional review boards in Paris.
Detailed clinical presentations of the patients are shown in Table 1. Briefly, all three patients (Pts 1, 2 and 3) were born to non-consanguineous parents (Figure 1A) and presented with disease within the first weeks of life. They all had developmental delay and axial hypotonia. Pts 1 and 2 developed spastic quadriplegia. Pts 2 and 3 developed pharmaco-resistant epileptic seizures, mainly myoclonic and associated with a marked photosensitivity in Pt 2; brain MRI showed variably severe ventriculomegaly (Pts 1, 2 and 3) and brain-stem and basal ganglia involvement (Pts 2 and 3) (Figure 1B).

(A) Segregation of the newly identified FARS2 variants into the pedigrees of the three reported families. (B) Axial T2-weighted images showing mild ventriculomegaly, no enlargement of the subarachnoid spaces in Pt 1; marked ventriculomegaly, enlargement of the subarachnoid spaces due to white matter loss, especially in the sylvian fissures, abnormal T2 hyperintensities in the lentiform nuclei and dorsal brainstem in Pt 2; moderate ventriculomegaly and enlargement of the subarachnoid spaces in Pt 3. (C) Linear map of the mtPheRS protein indicating its domain organization and position of the identified mutations. Evolutionary conservation of the amino acids affected by the newly described missense variants is shown in a multisequence alignment of mtPheRS proteins from different species.
Pathogenic variants were identified using a next generation sequencing panel targeting 382 genes involved in mitochondrial diseases (Pts1 and 3) or whole exome sequencing (Pt 2). The variants were assessed in silico, confirmed by Sanger sequencing and segregation analysis as in (15).
Skin fibroblast cultures from controls and patients, protein extraction and immunoblotting were carried out as in (16). MtPheRS antibody was purchased from Proteintech (Manchester, UK, 16436-1-AP). The remaining antisera used here were described previously (16).
Results
All three patients carried compound heterozygous variants including four missense mutations c.1256G>A p.(Arg419His), c.989G>A p.(Arg330His), c.1113G>T p.(Leu371Phe) and c.251A>C p.(His84Pro) all affecting evolutionary conserved amino acids and predictably pathogenic, and one duplication c.1269_1276dup p.(Ser426*) (Figure 1A and B, and Supplemental Table S1). None of these variants, but c.1256G>A, were previously reported in the literature.
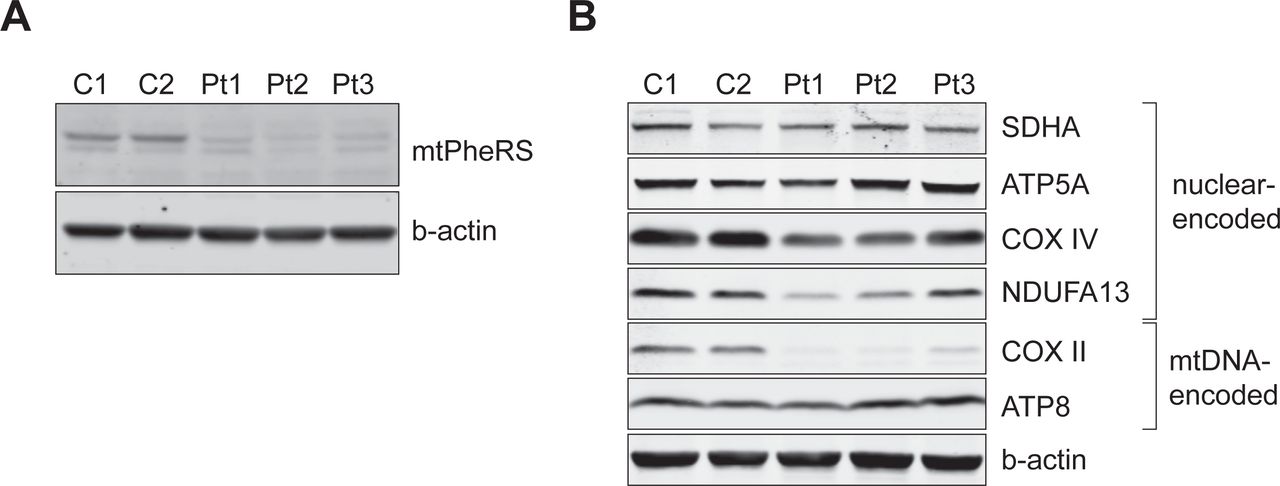
Immunoblotting of whole-cell extracts revealed a decreased abundance of mutant mtPheRS in all three patients’ fibroblasts suggesting a structurally destabilizing role of the identified mutations (Figure 2A). Examination of the abundance of mtDNA and nuclear-encoded OXPHOS proteins revealed a pattern of changes in their abundance consistent with impaired mitochondrial translation—decreased abundance of the mtDNA-encoded COXII protein and some, but not all, nuclear-encoded OXPHOS proteins (16), revealing multiple OXPHOS deficiency where CI and CIV are affected (Figure 2B). Interestingly, unlike COXII, the abundance of the mtDNA-encoded ATP8 was unchanged, probably because it contains a single phenylalanine whereas COXII contains ten (Figure 2B).
{kind=link}
{kind=link}
(A) Immunoblot analysis of whole-cell extracts from control (C1 and C2) and patient (Pt 1, Pt 2, Pt 3) fibroblasts showing decreased abundance of mtPheRS. β-actin was used as a loading control. (B) Immunoblot analysis of whole-cell extracts from control (C1 and C2) and patient (Pt 1, Pt 2, Pt 3) fibroblasts showing decreased abundance of mtDNA-encoded COXII (OXPHOS complex IV) and nuclear-encoded NDUFA13 (OXPHOS complex I) and COXIV (OXPHOS complex IV). Abundance of SDHA (OXPHOS complex II), ATP5A (OXPHOS complex V) and the mtDNA-encoded subunit ATP8 was unchanged. β-actin was used as a loading control.
Discussion
Our report expands the genetic spectrum of FARS2-linked pathologies with four novel pathogenic variants whose causal role was supported by bioinformatics and biochemical analyses. Clinically, these mutations conform to previously established presentations of FARS2 mutations. Early-onset epileptic phenotype—typically myoclonic epilepsy—is commonly seen in FARS2 patients and is invariably associated with developmental delay and a wide range of brain abnormalities. Most patients with this phenotype died before two years of age whereas two patients with juvenile onset epilepsy died soon after it became refractory (11). Two of the patients reported here, Pts 2 and 3, also presented an early-onset epilepsy, with mainly myoclonic pharmaco-resistant seizures, but were alive at the age of 16 and 5 years, respectively, suggesting that in some cases the early-epileptic phenotype can be associated with non-fatal disease. This is similar to another FARS2 patient reported in the literature who presented with generalized epilepsy at age of 19 months and was alive at 17 years of age (9).
Even though mutations in different mitochondrial tRNA synthetases consistently result in neurological phenotypes, these are generally not specific to individual enzymes including FARS2—early-onset epilepsy and HSP are genetically heterogeneous and not necessarily linked to tRNA aminoacylation. Moreover, there is no apparent correlation between the location of different FARS2 mutations along the mtPheRS polypeptide and the type, onset and severity of disease as, for example, mutations affecting the catalytic domain are just as likely to cause an epileptic phenotype as mutations affecting the ABD domain. This prevents us from establishing precise genotype-phenotype correlations. Most likely, the specific structural and functional effects of each mutation intersect with the genetic background to determine the extent to which tRNAPhe aminoacylation is impacted in each individual and the specific clinical presentation in that individual. For example, of the variants reported here, p.(Arg419His) was previously identified in a patient with pediatric spastic paraplegia and encephalopathy (17). Like this patient, Pt 1 had a non-fatal disease and did not present any epileptic phenotype. Although similar, the two phenotypes were not identical, which may result from the fact that the patient reported by Jou and colleagues was hemizygous whereas Pt 1 is compound heterozygous wherein p.(Arg419His) is co-expressed with a C-terminally truncated mtPheRS. Pt 3 also expressed the p.(Arg419His) variant but deviated from the other two patients in that it also had epileptic phenotype and white-matter degeneration resulting in a relatively more severe disease. In this case, too, the p.(Arg419His) variant is co-expressed with another mtPheRS mutant, p.(His84Pro), which may contribute to the specific phenotype observed in the patient. Thus, phenotypic differences in these cases may result from different genetic modifiers and/or the fact that in Pts 1 and 3 the p.(Arg419His) mutation is co-expressed with two other variants that may functionally interact during aminoacylation. Keeping in mind the small number of patients, it appears that the p.(Arg419His) variant is associated with non-fatal disease with or without epilepsy and white matter abnormalities. In the absence of in vitro activity data, we speculate that even though Arg419 is a structurally-important amino acid, its exchange to proline results in a semi-functional enzyme in manner similar to that proposed for p.(Arg419Cys) which resulted in spastic paraplegia and only transient epileptic seizures (12,18).
Our data further expands the genetic spectrum of FARS2 mutations and rises awareness that some patients may present with non-fatal early-onset epilepsy. As many patients with FARS2 mutations are compound heterozygous for two missense mutations it would be interesting to integrate data from in vitro activity assays of individual and combined mutant mtPheRS to determine to what extent, or if at all, different mutant mtPheRS interact functionally and whether this could add additional information that can help explain the phenotypic presentations observed in different patients.
Data Availability
Data are available upon request. Variants were submitted to ClinVar
Conflict of Interest
The authors have no competing financial interests
Funding
Grant Numbers: AFM-TELETHON, nr. 19876, nr. 22529, nr. 22251; ANR, GENOMIT ANR-15-RAR3-0012-07
Acknowledgements
We thank the patients and their families for participating in this study.
References




